

 science-review.ru
science-review.ru
Scientific journal
Scientific Review. Biological science
ISSN 2500-3399
ПИ №ФС77-57454
BIOLOGICAL ROLE AND PRACTICAL ASPECTS OF DIFFERENT TYPES OF DEOXYRIBONUCLEIC ACID METHYLATION AND DEMETHYLATION

Введение
У разных групп организмов основной мишенью для метилирования является цитозин, доля модификации которого варьирует от 3 до 30 %. Между тем до 3 % аденина в геномах представителей разных таксономических рангов могут также подвергаться подобному преобразованию. Таким образом, очевидно, что клетки эукариот и прокариот имеют по меньшей мере две системы ферментативного метилирования нуклеиновых кислот (аденин- и цитозинспецифическую) и специальные механизмы, регулирующие функции генов через комбинаторную логику подобных модификаций генома [1]. В основе подобных механизмов лежит влияние на конформацию хроматина и экспрессию генов за счет изменения эффективности узнавания соответствующих последовательностей дезоксирибонуклеиновых кислот (ДНК) ферментами нуклеинового обмена, гистоноподобными белками и различными факторами транскрипции.
Метилирование может происходить как в смысловых, так и в некодирующих участках генома и характеризовать совершенно различные процессы. К смысловым относят участки, несущие информацию о последовательности аминокислот структурно-функциональных белков организма (экзоны генов), в то время как некодирующими можно назвать цис- и трансрегуляторные элементы, интроны, ретротранспозоны, вирусные элементы, псевдогены, теломеры, тандемные повторы и т.п. Однако некодирующие участки также очень разнородны, поскольку среди них имеются как последовательности, непосредственно влияющие на процессы транскрипции и трансляции, так и интактные области так называемой «мусорной» ДНК.
Цель исследования – анализ современного взгляда на молекулярно-генетические характеристики и выявление актуальных научно-практических тенденций в области изучения процессов метилирования и деметилирования наследственного материала.
Задачи исследования: анализ современных взглядов на биохимические основы процесса метилирования и деметилирования нуклеиновых кислот, включающие формы метилтрансфераз и их субстратную специфичность, изучение информации, касающейся поддерживающей и спонтанной форм метилирования, определение общебиологического и биомедицинского значения разных вариантов метилирования и деметилирования генов и их промотерных регионов.
Материалы и методы исследования
В основу работы положен комплекс обширных теоретических исследований. С сентября 2024 г. по апрель 2025 г. проводился поиск и анализ научно-практической литературы по исследуемой теме. Всего для работы над статьей на цифровых международных и отечественных платформах (Научная электронная библиотека elibrary.ru, PubMed, Web of Science, Scopus и др.) изучено и проанализировано, охватывая период с 1990 по 2025 г., более 460 научных трудов, из которых 50 наиболее актуальных и информативных были использованы в настоящей работе.
Результаты исследования и их обсуждение
Биохимические основы метилирования нуклеиновых кислот
Метилирование ДНК представляет собой обратимую ковалентную реакцию ферментативного переноса метильной группы с донора S-аденозил-L-метионина на акцепторы: C-5 или N-4 положения цитозина и N-6 положение аденина (рис. 1). Процесс катализируется сложными ферментами, известными как ДНК-метилтрансферазы или ДНК-метилазы (DNA MTase, DNMT) [2, 3].
В противовес метилазам действуют ферменты, называемые деметилазами. К основным участникам элиминирования метильной группы относятся белки семейства TET- диоксигеназ (Ten-Eleven-Translocation), превращающие 5-метилцитозин (5mC) в 5-гидроксиметилцитозин (5hmC), за счет добавления гидроксила по пятому положению пиримидинового кольца. Замена окисленных производных 5hmC на обычный цитозин происходит при участии фермента тимин-ДНК-гликозилазы (TDG). Наиболее часто 5hmC и его окисленные производные, как промежуточные стадии деметилирования, возникают у вирусов, в клетках некоторых структур мозга и в эмбриональных клетках позвоночных [4, 5]. В аналогичных реакциях деметилирования N6-метиладенина (6mA) участвуют ферменты ДНК-метиладениндеметилазы (DMAD), ортологичные семейству ТЕТ [6].
По отношению к субстрату ДНК-метилазы могут быть разделены на три группы:
‒ Dam (EC 2.1.1.72) – специфичные к N6-положению аденина;
‒ N4-Dcm (EC 2.1.1.113) – специфичные к N4-положению цитозина;
‒ C5-Dcm (EC 2.1.1.37) – специфичные к C5-положению цитозина.
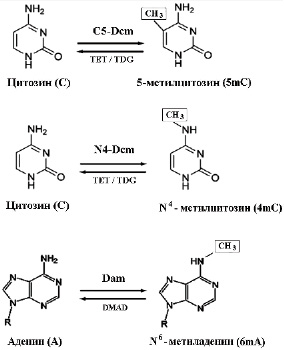
Рис. 1. Варианты метилирования ДНК, распространенные в живой природе Источник: составлено авторами
Справедливости ради, первые две группы нельзя назвать классическими метилазами. В бóльшей степени они могут быть отнесены к ферментам системы рестрикции-модификации, проявляющим сопутствующую метилтрансферазную активность.
Активность метилтрансфераз в клетках прокариот и эукариот, как и в клетках зрелых, эмбриональных и малигнизированных тканей может существенным образом отличаться. Метилирование при этом редко носит спонтанный, ненаправленный характер. Например, Dam, с высокой степенью специфичности, узнают аденин в последовательностях 5’-GpApUpC-3’, 5’-GpGpApU-3’ и 5’-UpRpUpApY-3’ (R = A/G; Y = C/U; p – фосфат) [7, 8] и способны к метилированию de novo, в то время как ферменты Dcm обычно метилируют цитозин в совокупностях нуклеотидов 5’-CpG-3’ и 5’- CpNpG -3’ (где N – любой нуклеотид) в поддерживающем режиме, от одного клеточного деления к другому. Хотя в целом ряде случаев такие гомологи Dcm, как DNMT1 и DNMT3A, демонстрируют способность к эффективному переносу метильных групп de novo [9]. Следует также отметить, что у растений описаны случаи, когда Dcm метилировали цитозин в нехарактерных динуклеотидах 5’-CpC-3’ , 5’-CpT-3’ и 5’-CpA-3’. Вопрос о присутствии подобного варианта метилирования у человека и животных долгое время относился к разряду контроверсий. Однако данные последних лет свидетельствуют о том, что такой тип метилирования встречается в нейронах и эмбриональных стволовых клетках млекопитающих, участвуя в дифференцировке плюрипотентных клеток и синаптогенезе [10].
До недавнего времени, считалось, что Dam и N4-Dcm – метилазы характерны только для прокариот, в то время как C5-Dcm-аналоги встречаются преимущественно в ядерных клетках. Но данные последних лет показали, что метилазы группы Dam присутствуют и у эукариот [7, 8].
По последним данным, 70 % всех сайтов CpG в ДНК соматических клеток млекопитающих метилированы. Стоит отметить, что здесь речь идет о постнатальном периоде развития. В период же эмбриогенеза эти цифры существенно отличаются. К постоянно высокометилированным последовательностям у взрослого организма можно отнести мобильные генетические элементы, сателлитную ДНК, межгенные участки и первые экзоны определенных генов.
Подавляющее большинство CpG-динуклеотидов распределено по геному в виде одиночных динуклеотидов, оставшаяся же часть формирует особые зоны скопления – CpG-островки.
В геномах эукариот встречаются скопления CpG, уровень метилирования которых значительно ниже и не столь стабилен, чем в геноме в целом. Установлено, что такие группы CpG, или островки, находятся в промоторных зонах более чем 60 % генов. CpG-островком в широком смысле называется участок ДНК длиной более 200 п.о. (пар оснований) с содержанием (C + G) ≥ 50 % и соотношением наблюдаемого числа CpG к произведению C и G на уровне более 0,6. CpG-островки обычно находятся в промоторной области и в первом экзоне белок-кодирующих генов млекопитающих, включая все гены домашнего хозяйства (англ. – housekeeping) и часть тканеспецифичных генов. Установлено, что в геноме человека присутствует более 45 тыс. CpG-островков [11]. Между тем одиночные динуклеотиды CpG, рассеянные по всему геному, также могут периодически метилироваться. В результате, если, в качестве примера, взять соматические клетки млекопитающих, то становится очевидно, что около 75 % всех CpG в них метилированы [12].
Сайленсинг участка генома при гиперметилировании в большинстве случаев обеспечивается не только метильным кэпом, но и метилцитозин-связывающими белками, которые способны сорбироваться на метилированных CpG-динуклеотидах. Такие белок-нуклеотидные комплексы привлекают деацетилазу гистонов (HDAC) и многочисленные факторы, участвующие в ремоделировании хроматина.
Пожалуй, наиболее перспективным предметом для исследований представляется метилирование и деметилирование цис-/трансрегуляторных элементов генов, определяющее непосредственное регулирование функционального статуса гена и уровня экспрессии кодируемого им продукта без изменения в кодирующей последовательности.
Общебиологическое значение метилирования ДНК
Как уже отмечалось, нормальное физиологическое метилирование может быть условно разделено на два базовых варианта:
‒ поддерживающее метилирование,
‒ спонтанное (de novo) метилирование.
Первый вариант в некотором роде обуславливает «эпигенетическую наследственность», то есть сохранение уже имеющегося эпигенотипа или, иначе, наследуемого паттерна метилирования. Данный рисунок, по сути, ответственен за видоспецифический пейзаж включенных и выключенных генов. Именно его нарушение может приводить к возникновению онкологических заболеваний, нечувствительности к тем или иным гормонам и биомедиаторам, развитию артефактных рудиментов и атавизмов и т.п.
Второй вариант, или спонтанное метилирование, обеспечивает «эпигенетическую изменчивость». Иначе говоря, уровень метилирования CpG в геноме имеет не только большое значение в сфере регулирования экспрессии генов, но также огромное эволюционное значение, поскольку оно увеличивает частоту спонтанных мутаций. Это связано с тем, что метилированный цитозин способен к дезаминированию с образованием остатков тимина (рис. 2). Динуклеотиды CpG в ряду поколений претерпевают транзицию в динуклеотиды TpG, что подтверждается недостаточной представленностью CpG в геномах эволюционно более поздних видов [13]. Устойчивая транзиция по охарактеризованному выше принципу, по всей видимости, происходит лишь в экстремальных условиях.
Известно, что дезаминирование неметилированного цитозина с образованием урацила часто встречается в ДНК погибших клеток. Прижизненное же дезаминирование внеостровкового 5-метилцитозина связывают с работой цинкзависимых цитидиндезаминаз класса APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) (рис. 2) (подробнее см. в разделе «Метилирование и инфекционный процесс»). Такие изменения происходят через стадии образования 5-гидроксиметилцитозина (5hmC) и 5-гидроксиметилурацила (5hmU) и могут завершиться появлением апиримидинового сайта (АП, АР), то есть участка в нуклеотидной последовательности, в котором прервана связь между остатком дезоксирибозы и пиримидиновым основанием, в то время как сахарофосфатный остов полностью сохранен.
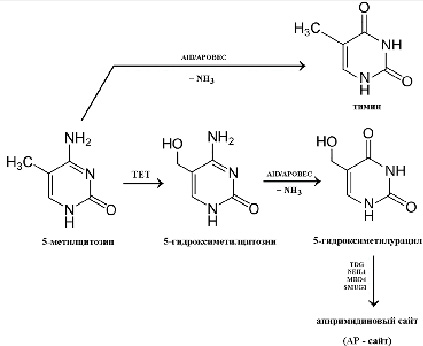
Рис. 2. Основные механизмы дезаминирования 5-метилцитозина Источник: составлено авторами
Данные изменения чаще возникают при активации внутриклеточной защиты от вирусной рибонуклеиновой кислоты (РНК) или ДНК, но если подобное изменение произошло в эукариотической ДНК, то такие модификации в норме достаточно быстро распознаются как опасные точковые мутации и нивелируются специальными внутриклеточными системами репарации, например с помощью фермента BER (base excision repair) [14].
Биомедицинское значение метилирования ДНК
Метилирование и инфекционный процесс
Известно, что при атаке различными патогенами как хозяйские клетки, так и сами инфекционные агенты зачастую претерпевают каскадные изменения в своих транскрипционной и эпигенетической программах, мобилизуя гены, необходимые для ключевых метаболических превращений.
Так, некоторые бактерии при внедрении в целевой многоклеточный организм начинают продуцировать активаторы транскрипции, мимикрирующие под эукариотические аналоги, направляя тем самым обменные процессы клеток хозяина в нужное им русло [15]. При этом не последнюю роль может играть сверхэкспрессия бактериальных метилаз, обуславливая формирование некоторых параметров вирулентности патогенных и условно-патогенных бактерий. Многие факторы транскрипции микробных клеток весьма чувствительны, например, к метилированию промотора papBA белка пилина [16]. Как известно, данный белок это не что иное, как основной структурный компонент фимбрий, обеспечивающих подвижность бактерий, хемотаксис, их проникновение в среды и ткани организма, образование биопленок и перенос генетического материала при конъюгации. Все перечисленные процессы относятся к мощным факторам вирулентности микроорганизмов.
Другое интересное наблюдение демонстрирует, что внедрение инфекционных агентов запускает деметилирование промоторных и энхансерных элементов уже в клетках хозяина, включая те из них, что регулируют активацию ключевых факторов транскрипции иммунных белков. Активная утрата метильных меток в геноме связана с общим эпигенетическим ремоделированием, включающим стойкое усиление меток активации ДНК-связывающих белков (метилирование остатков лизина в гистонах), меток деметилирования ДНК (5hmC) и в конечном счете увеличение стерической доступности целевых генов в хроматине иммунных клеток. Очевидно, что подобные изменения метилирования гистонов и ДНК в энхансерных участках играют ранее недооцененную роль эпигенетической иммунной памяти при регуляции прицельного транскрипционного ответа на инфекцию даже в непролиферирующих клетках [17].
Реакции метилирования и деметилирования также чрезвычайно важны для сопротивляемости организма внедрению и активации вирусных агентов. Важную роль здесь играет ранее упомянутая цитидиндезаминаза APOBEC3G – часть большой системы адаптивного внутриклеточного иммунитета, направленного на ингибирование репликации вирусов и мобильных генетических элементов в эукариотическом геноме. Активность подобных ферментов проявляется, прежде всего, в узнавании и дезаминировании метилированного цитозина в урацил (C→U) в вирусной (-) цепи ДНК во время обратной транскрипции РНК-матрицы, что, в свою очередь, опосредует точковую мутацию гуанозина в аденозин (G→A) в (+) цепи ДНК, несущей информацию о функциональных белках вируса. Положительным моментом здесь является то, что некоторые из данных мутаций могут носить инактивирующий характер по отношению к вирусам или ретротранспозонам [18].
Метилирование ДНК при метаболических нарушениях
Метилирование играет важную роль в управлении работой ключевых генов, вовлеченных в обеспечение гомеостаза функциональных метаболитов клеток человека и животных. Например, метилирование промотора гена инсулина усиливается у пациентов с сахарным диабетом 2 типа (T2Д) и отрицательно коррелирует с экспрессией гена инсулина в островках Лангерганса поджелудочной железы [19]. Нарушения нормального баланса метилирования регуляторных элементов генов глюкозного транспортера 4-го типа (GLUT4) и рецепторов, активируемых пероксисомными пролифераторами PPAR-γ-2, также обнаруживаются в этиологии возникновения инсулиновой резистентности и последующего развития T2Д [10].
Международной исследовательской группой под руководством S. Wahl на примере прогнозирования развития T2Д показано, что по характеру изменения метилирования можно предсказать риск развития этого патологического состояния, причем с большей уверенностью, чем по таким традиционным маркерам, как тип ожирения или содержание глюкозы в крови [20].
Метилирование ДНК выступает также одним из основных эпигенетических механизмов, играющих важную роль в инициации и распространении атеросклероза [21]. Имеются свидетельства о том, что смещение баланса выработки инсулина организмом в сторону его увеличения опосредованно способствует сверхэкспрессии некоторых разновидностей ДНК-метилтрансфераз, индуцирующих гиперметилирование второй экзонной области гена рецептора к эстрогену ER-α. Это снижает экспрессию ER-α, тем самым нарушая гормональную регуляцию нормальной пролиферации гладкомышечных клеток кровеносных сосудов, что лежит в основе развития атеросклеротических изменений стенок сосудов и капилляров [22].
Участие метилирования ДНК в развитии онкологических заболеваний
В качестве одной из причин возникновения целого ряда генетических заболеваний и злокачественных новообразований называется сбой эпигенетической программы регуляции активности генов, основанной на аномальных изменениях метилирования СpG-островков в регуляторных регионах, что приводит к их гиперактивации или полной инактивации [23].
В основе неопластических трансформаций могут лежать не только нарушения метилирования цитозина, определенную роль в этом процессе, по всей видимости, способно сыграть также аномальное метилирование остатков аденина в мРНК. Так, научные группы Wu и Meyer в ходе изучения большой выборки смысловых и интактных последовательностей геномной ДНК, а также мРНК пришли к выводу о том, что m6A сайтами в организме некоторых эукариот обогащены преимущественно ретротранспозоны, 3′-нетранслируемые области и последовательности, располагающиеся перед стоп-кодонами некоторых генов [24, 25]. Результаты последующих исследований показали, что специфическое ингибирование посттрансляционного метилирования аденина за счет сайленсинга N6-аденозин-метилтрансферазы (METTL3) – одного из важнейших представителей группы Dam – достаточно для продления циркадных ритмов и замедления созревания первичного транскрипта, что есть необходимое условие для снижения пролиферативной активности различных клеток [26].
До недавнего времени бытовало мнение о том, что в основе развития большинства опухолей лежит гипометилирование протоонкогенов. Данные последних лет исключают столь однозначную интерпретацию. Многие события, связанные с деметилированием генов и их регуляторных областей, могут встречаться в совершенно здоровых клетках как молодых, так и стареющих организмов [27]. Но нельзя отрицать то, что гипометилирование приводит к изменению транскрипционной активности многих онкогенов. Взаимосвязь между гипометилированием и изменением экспрессии была показана для инсулиноподобного фактора роста второго типа, длинных диспергированных повторов LINE1, Alu элементов типа Yb8, центромерных повторов Sat-α и тандемных повторов NBL-2 при колоректальном раке, раке желудка и мочевого пузыря. В отличие от этого, при большинстве лейкозов не наблюдается существенных изменений в паттерне метилирования LINE1 и Alu Yb8, хотя метилирование NBL-2 и субтеломерных повторов D4Z4 может несколько возрастать [28].
Важным в развитии тех или иных опухолевых процессов выглядит понятие «горячих точек» метилирования, то есть локальных зон повышенной эпигенетической нестабильности. Например, участок хромосомы 11p – это известная «горячая точка» гиперметилирования CpG-островков в солидных опухолях, лейкемиях и вирус-ассоциированных опухолях [11].
Некоторые разновидности рака, включая рак желудка, характеризуются гиперметилированием генов-онкосупрессоров, таких как CTNNB1, RASSF1A, APC и SFRP1 и генов-регуляторов транскрипции и клеточного цикла (WT1, CDKN2B, CDKN2A, и PRDM2), а также промоторов некоторых компонентов канонического сигнального пути Wnt/β-катенин [29]. Например, результатом метилирования промоторного региона гена DKK-3 (антагонист Wnt) становится избыточная активация Wnt-пути, нарушающая клеточную пролиферацию и дифференцировку, приводя к малигнизации [30].
Накопление β-катенина в клетках вследствие сбоя в регуляции данного сигнального пути может играть одну из ключевых ролей в развитии злокачественных образований печени, толстой кишки, поджелудочной железы, эндометрия, а также опухолях околощитовидных желез у пациентов со вторичным гиперпаратиреозом [31, 32].
С другой стороны, гиперметилирование – также не всегда надежный маркер онкогенеза. Обширные экспериментальные данные указывают, например, на то, что до 90 % генов, якобы гиперметилирующихся de novo при некоторых раковых заболеваниях, на самом деле уже конститутивно репрессированы в нормальных тканях. Помимо этого, ряд неоплазий детерминирован исключительно лишь сочетанием гиперметилирования однокопийных генов онкосупрессоров и гипометилирования тандемных повторов [11, 33].
Следует особо отметить, что порой сам факт метилирования не выступает в качестве решающего при развитии той или иной патологии. В ряде случаев переход к негативному сценарию может быть обусловлен возрастающей вероятностью транзиций в динуклеотидах CpG по причине спонтанного дезаминирования 5-метилцитозина при воздействии солей тяжелых металлов, радиации, нарушении работы цитидиндезаминаз класса APOBEC и т.д. Например, в случае опухолей ассоциированных с изменением экспрессии онкосупрессора р53, частота подобных точковых мутаций достигает 25–50 % [11, 34].
Таким образом, среди значимых процессов, с помощью которых метилирование может участвовать в формировании онкогенного фенотипа, целесообразно выделить следующие:
‒ нарушение работы ДНК-метилтранс-фераз и цитидиндезаминаз (APOBEC),
‒ гипометилирование регуляторных элементов онкогенов и тандемных повторов,
‒ гиперметилирование регуляторных элементов онкосупрессоров,
‒ спонтанные точковые мутации вследствие дезаминирования CpG-динуклеотидов [11, 35].
Метилирование ДНК в неврологии и психиатрии
В аспекте проблематики настоящего обзора также привлекают внимание работы, касающиеся эпигенетических следов, лежащих в основе пластичности функционирования нервных клеток и их контактов, способствующей постоянному влиянию жизненного опыта на поведение и физиологию индивида, начиная от формирования долговременной памяти и заканчивая последствиями травм или приема психотропных препаратов [36].
Аномалии глобального метилирования хроматина, отдельных генов и регуляторных элементов могут играть определенную роль в познавательных способностях, формировании памяти и в этиологии развития многих форм психических заболеваний.
Динамика метилирования ДНК или ДНК-связывающих белков вызывает устойчивые изменения в паттернах экспрессии нескольких генов, вовлеченных в синаптическую пластичность, с помощью которой реализуется феномен памяти и обучения. К этим генам относятся гены нейротропного фактора мозга (BDNF), кальциневрина, протеиновой фосфатазы 1 (PP1) и гликопротеина рилина [37].
Указанные гены и их белковые продукты – важные терапевтические мишени при лечении различных заболеваний ЦНС. Так, показано, что включение L-метионина в схему лечения больных шизофренией для улучшения работы метаболизирующих дофамин ферментов, наоборот, обуславливает неожиданное обострение состояния таких пациентов. Дальнейшие исследования продемонстрировали, что подобный рецидив был связан с изменением экспрессии гена белка рилина, колебания уровня которого коррелируют со степенью выраженности психотических проявлений при шизофрении, биполярном расстройстве и аутизме. В результате изучения был получен материал, анализ которого позволил заключить, что подобные колебания экспрессии рилина определенным образом связаны с гиперметилированием CpG островков промоторной области его гена в позициях между -139 и -134 [38].
Метилирование ДНК в судебной медицине
К исторически первым методам молекулярно-генетической дактилоскопии можно отнести метод ДНК-профилирования по мини- и микросателлитным последовательностям. Он стал незаменимым инструментом для специалистов, занимающихся расследованием различных преступлений и юридических споров. Данный метод имеет ограничения, основным из которых является присутствующая вероятность полного совпадения ДНК родных (не однояйцовых) братьев и сестер, составляющая 1:100000. Однако последовательности нуклеотидов в ДНК однояйцовых близнецов абсолютно идентичны. Это обстоятельство затрудняет работу криминалистов и судебных экспертов, работающих над выяснениями обстоятельств преступлений, в которых были замешаны близкие родственники или близнецы, что сопряжено с риском вынесения ошибочных оправдательных или обвинительных судебных решений.
Поэтому в последние годы в разных странах активизировались работы, направленные на преодоление указанных ограничений [39, 40]. Методы идентификации ДНК близнецов разрабатывались и ранее. Самый точный из них сводится к полному секвенированию геномов обоих близнецов для идентификации мутаций, которые могли появиться в геноме одного из них. Если такая мутация обнаруживается, в собранном на месте преступления биологическом материале проводится ее прицельный поиск. Этот подход зачастую оказывается неприемлемым в реальных условиях работы, поскольку требует больших временных и финансовых затрат [40].
Прицельное метиломное профилирование кодирующих и некодирующих регионов ДНК представляет собой значительный прогресс в разработке сравнительно дешевого и быстрого метода выявления различий даже между генетически идентичными близнецами.
Поскольку на метилирование в раннем возрасте оказывают влияние не только внутренние особенности, но и некоторые факторы окружающей среды (питание, курение, радиационный фон и т.п.), поэтому изучение рисунка метилирования маркерных локусов ДНК, пожалуй, единственный метод, который потенциально способен отличить биоматериалы однояйцовых близнецов и даже предсказать те условия, в которых происходило становление их организма.
В ряде работ также было показано, что, например, промоторы некоторых генов стабильно наследуют отцовский, а не материнский рисунок метилирования, что говорит о наличии такого понятия, как отцовский метиломный импритинг, который также весьма информативен с точки зрения «молекулярной дактилоскопии». Многочисленные исследования последнего десятилетия показывают, например, возможность определения пола посредством изучения метилирования макросателлитной последовательности DXZ4, ассоциированной с X-хромосомой [41]. Продемонстрировано, что данный ген гипометилирован на импритированной Х-хромосоме и гиперметилирован на активной Х-хромосоме. Также показана возможность определения пола на основании изучения статуса метилирования гена HOXA4 в буккальном эпителии человеческой слюны. Данный ген был гиперметилирован у мужчин и преимущественно гипометилирован у женщин [42].
Метилирование ДНК и старение
Многочисленные исследования демонстрируют отчетливую коррелятивную связь между метилированием ДНК, старением и возрастными заболеваниями [43]. Атипичные паттерны метилирования регистрируются в смысловых и интактных последовательностях человеческого генома при физиологическом увядании и при таких возрастных патологиях, как болезнь Альцгеймера, паркинсонизм, остеоартроз и многих других [44, 45].
Не раз было продемонстрировано изменение метилирования повторов Alu, сопровождавшее возрастные изменения, причем статистически наиболее достоверное снижение уровня метилирования наблюдалось в интервале 34–68 лет [46]. Усиление регуляции ретропозиции Alu элементов при деметилировании прежде всего обуславливает ядерную цитотоксичность, которая связана с образованием стойких очагов повреждения ДНК и утратой способности к эффективной репарации в перицентрическом районе хромосом. Цитотоксичность активированных повторов имеет особое значение при старении стволовых клеток у взрослого человека. Поэтому подавление транскрипции Alu может восстановить пластичность клеток и способность их самообновления за счет воздействия на экспрессию так называемых «главных» регуляторов плюрипотентности (Nanog, Oct4 и др.) [47].
С точки зрения связи эпигенетических изменений с гериатрией особенно интересны наблюдения, проводившиеся за группами лабораторных мышей. Одна группа была рождена от старых самцов (>120 недель), в то время как другая – от молодых (< 120 недель). Проведенное тестирование показало, что потомки наследуют элементы метиломного эпигенетического пейзажа отцов. При этом у мышей, рожденных от старых отцов, наблюдались нарушения в условнорефлекторных реакциях пассивного избегания, достоверное снижение продолжительности жизни, задержка активности и сенсомоторного развития по сравнению с мышами, рожденными от более молодых отцов. В то же время двигательная активность потомков и их способность к обучению были также значительно снижены у мышей, рожденных от очень молодых отцов (в возрасте 6 недель), по сравнению с теми, кто родился от отцов нормального возраста (≥ 12 недель) [48].
За последнее десятилетие в области геронтологии и гериатрии наметился существенный прогресс. Накопленный массив данных, касающихся возрастной динамики эпигенетических изменений, происходящих в различных участках генома, позволил выявить панель локусов, по рисунку метилирования которых с высокой точностью можно определить даже хронологический возраст человека [49]. При этом фактически было введено новое понятие «эпигенетических часов». Самым известным примером таковых хочется назвать часы Хорвата, представляющие, по сути, весьма точную методику оценки хронологического возраста, учитывающую 353 эпигенетических СpG-маркера в человеческом геноме, ставших фактически часовыми стрелками этого «механизма» [50]. На основании данной методики Хорват установил импровизированный «циферблат», включающий четыре основных критерия метиломного возраста:
‒ во-первых, он близок к нулю в эмбриональных и плюрипотентных стволовых клетках;
‒ во-вторых, он коррелирует с номером пассажа клеток;
‒ в-третьих, он становится более релевантным в более позднем возрасте;
‒ в-четвертых, метод применим к тканям высших приматов, таким как, например, шимпанзе (которые используются в качестве модельных объектов в подобных исследованиях).
С каждым годом база данных по метиломным маркерам пополняется новыми локусами и паттернами, поэтому в «эпигенетических часах» со временем обязательно должны будут появиться «минутные» и даже «секундные» стрелки.
Заключение
Накоплено немало фактов, свидетельствующих о большом вкладе дефектов реакций метилирования и деметилирования в возникновение, развитие и исход многих патофизиологических состояний. При этом возникает необходимость систематизации и осмысления этой ценной информации. Анализ имеющейся литературы показывает наличие общей тенденции, заключающейся в стремлении привести логику метилирования к некоему простому правилу, гласящему, что, например, условием для развития возрастных заболеваний и подавляющего большинства опухолей служит сочетание гиперметилирования регуляторных элементов с общим гипометилированием остальных участков генома. Необходимо констатировать тот факт, что до сих пор не существует надежного правила касательно того, что же, собственно, является краеугольным камнем в структуре развития того или иного отклонения от нормы: гиперметилирование, гипометилирование либо какие-то их сочетания. Имеется множество примеров заболеваний, ассоциированных как с высоким, так и с низким уровнем метилирования, когда перенос метильных групп может оказывать не только активирующее, но и сайленсирующее влияние.
Между тем в большинстве случаев паттерны метилирования геномной ДНК в норме и при патологии существенным образом различаются. Наиболее достоверную выраженность эти различия принимают в случае изменения метилирования цис- и трансрегуляторных элементов, таких как промотеры, энхансеры, 3′-нетранслируемые области, транспозоны и т.п.
По всей видимости, в данном случае не последнюю роль должно играть и то, какие функциональные белки в конечном счете активируются или, наоборот, дезактивируются при подобном воздействии (онкогены или онкосупрессоры), влияя на пролиферацию, способность к аресту клеточного цикла или на способность клеток вступать в апоптоз.
Последовательная расшифровка «эпигенетического кода», несомненно, должна стать не менее важной вехой в биомедицинской науке, чем была расшифровка кода генетического. Это в перспективе позволит предоставлять более точный прогноз на ранних стадиях развития заболеваний, осуществлять эффективную диагностику и разрабатывать адекватные схемы лечения.
Библиографическая ссылка
Касап Е.Ю., Парфенова О.К., Сидоров Н.Г., Гришин Д.В. БИОЛОГИЧЕСКАЯ РОЛЬ И ПРИКЛАДНЫЕ АСПЕКТЫ РАЗЛИЧНЫХ ВАРИАНТОВ МЕТИЛИРОВАНИЯ И ДЕМЕТИЛИРОВАНИЯ ДЕЗОКСИРИБОНУКЛЕИНОВЫХ КИСЛОТ // Научное обозрение. Биологические науки. 2025. № 2. С. 70-80;URL: https://science-biology.ru/en/article/view?id=1409 (дата обращения: 15.07.2026).
DOI: https://doi.org/10.17513/srbs.1409